Retinitis pigmentosa er en gruppe af arvelige sygdomme i øjets nethinde. Disse sygdomme har samme udseende og giver de samme symptomer
Personer med retinitis pigmentosa starter med at få besvær med at orientere sig i mørke, hvorefter der kommer indsnævring af synsfeltet. Det skarpe syn i midten af synsfeltet bliver ramt til sidst
Det er for nylig blevet muligt at behandle en enkelt af de mange hundrede kendte arvetilstande med retinitis pigmentosa ved hjælp af genterapi, og det forventes at flere arvetilstande bliver tilgængelige for genterapi i fremtiden
Sygdommens skadelige effekter på synet kan i mange tilfælde afhjælpes eller lettes med optiske hjælpemidler
Hvad er retinitis pigmentosa?
Retinitis pigmentosa er en gruppe af arvelige sygdomme i øjets nethinde med fremadskridende tab af synet. Retinitis pigmentosa kan være forbundet med misdannelser i andre af kroppens organer.
Hvad er symptomerne på retinitis pigmentosa?
De første symptomer på retinitis pigmentosa er tiltagende besvær med at færdes i mørke. Senere kommer der indsnævring af synsfeltet, på samme måde som hvis man ser gennem et rør (kikkertsyn). Til sidst kan også det centrale syn blive påvirket.
Hvordan stilles diagnosen?
Når sygdommen er kendt i familien, kan øjenlægen undersøge, om andre familiemedlemmer har de tidlige stadier af sygdommen.
Hvis man har nedsat nattesyn, kan øjenlægen ved at undersøge synsfeltet mere detaljeret, finde udfald i dette, mens synsstyrken ofte kan være normal i meget lang tid.
Hvis øjenlæge har mistanke om retinitis pigmentosa, bliver man henvist til en øjenafdeling, som er specialiseret i retinitis pigmentosa. Man vil normalt få undersøgt sin synsstyrke, synsfelt og øjets evne til at tilpasse sig mørke.
Man får også foretaget en elektroretinografi (forkortet ERG). Dette er en undersøgelse, som måler nethindens funktion. ERG bruger øjenlægen til at:
Stille en præcis diagnose
Vurdere i hvilken grad synet er påvirket
Følge sygdommens forløb og på den måde kunne sige noget om prognosen
Vurdere effekten af behandling
Endelig får man tilbud om at få taget en blodprøve, som sendes til undersøgelse for at finde ud af, om man har en af de former for retinitis pigmentosa, hvor gendefekten kendes. Dette giver dels bedre muligheder for at informere om sygdommens udvikling. Det betyder også, at man kan kontaktes, hvis der kommer en behandling for den type gendefekt, man har.
Hvorfor får man retinitis pigmentosa?
Sygdommen er arvelig og kan nedarves som
En såkaldt dominant sygdom, hvor der er 50% risiko for at videregive sygdommen
En såkaldt recessiv sygdom, hvor man oftest ikke kender andre, der har sygdommen i familien
En kønsbunden sygdom, hvor det typisk er drenge der rammes, og hvor sygdommen er nedarvet gennem deres mor
Retinitis pigmentosa er som regel begrænset til øjet. 20-30% af de der har retinitis pigmentosa, har andre sygdomme samtidig andre steder i kroppen, og disse tilfælde indgår i mere end 30 forskellige syndromer. Den aftagende funktion af nethinden skyldes et tiltagende tab af de lysfølsomme sanseceller (stave og tappe).
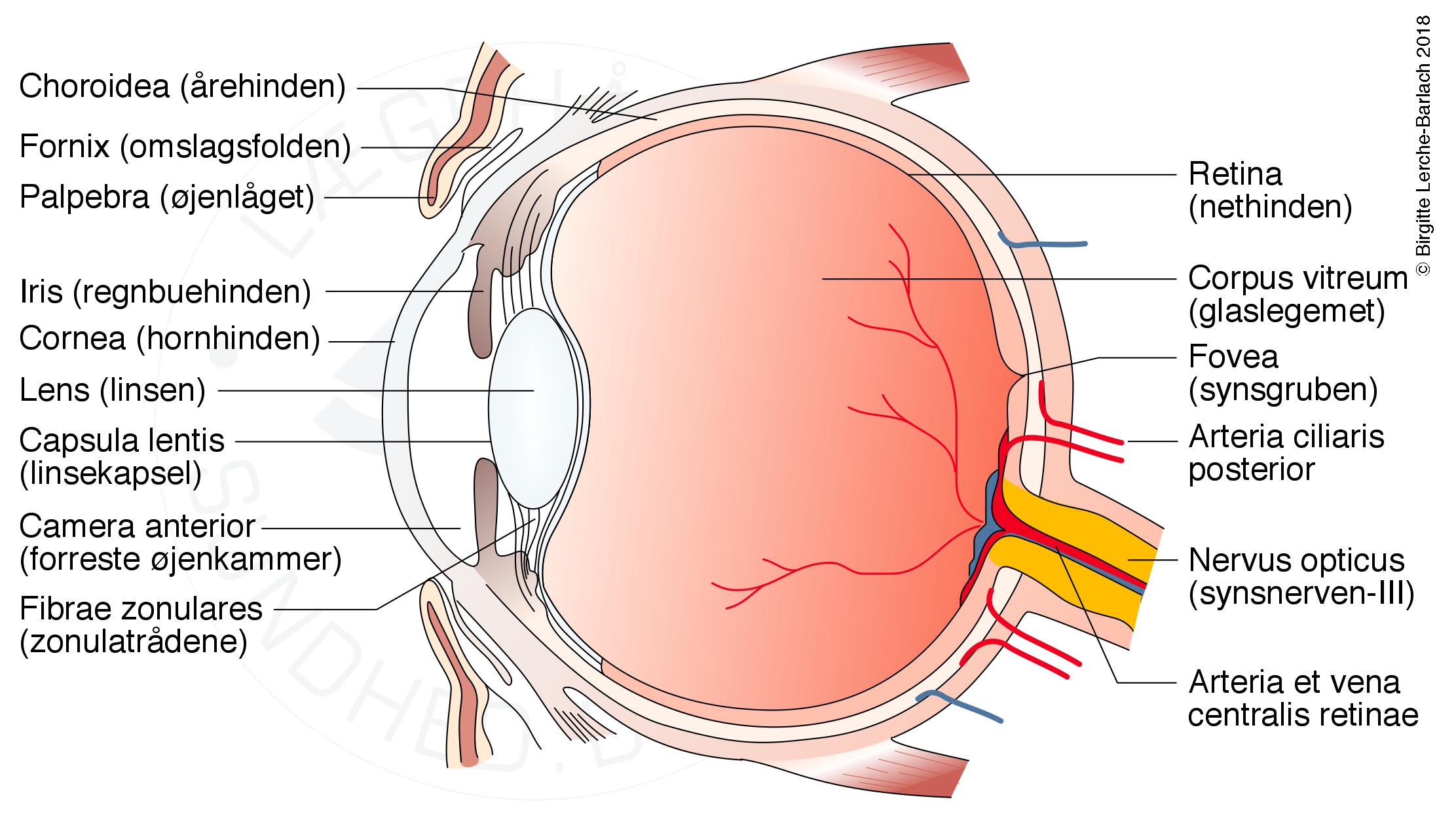
Øjet, tværsnit
Forskellig syndromer forbundet med retinitis pigmentosa
Et syndrom er betegnelsen for en samling af flere tegn på sygdom, symptomer, eller karakteristika som ofte forekommer sammen og karakteriserer en bestemt sygdom.
Ushers syndrom
Ved Ushers syndrom er retinitis pigmentosa forbundet med nedsat hørelse. Dette er den hyppigste form for retinitis pigmentosa som led i et syndrom. Dette syndrom udgør omkring 20-40% af alle personer med såkaldt recessiv sygdom.
Høretabet kan være udtalt og til stede fra fødslen. Det kan være forbundet med balanceforstyrrelser (Ushers syndrom type I) eller være moderat uden tendens til forværring (type II). Hørelsen kan også være normal i barndommen og ungdommen, hvorfor man med tiden kan få et betydeligt høretab (type III).
Ushers syndrom kan skyldes ændringer i mindst 11 gener. De forskellige ændringer i arvematerialet (mutationer) i disse gener fører til forskelle i symptomerne.
Bardet-Biedl syndrom
Ved Bardet-Biedl syndrom er retinitis pigmentosa i varierende grad forbundet med fedme, mental retardering, ekstra fingre (polydaktyli), manglende kønsudvikling (hypogonadisme) og sygdom i nyrerne. Nogle får nyresvigt og har behov for nyretransplantation. Dette syndrom udgør 5-6% af tilfældene med retinitis pigmentosa.
Andre syndromer
Af de mange syndromer, som omfatter retinitis pigmentosa, er tre former særligt vigtige, fordi tidlig behandling kan bevare synet. Det gælder abetalipoproteinemi (Bassen-Kornzweig syndrom), phytanisk oxygenoksidase mangel (Refsums sygdom) og familiær isoleret vitamin E mangel.
Hvordan behandler man retinitis pigmentosa?
Der er nyligt udviklet en behandling med genterapi til den arveform, der kaldes RPE65 associeret retinitis pigmentosa. Behandlingen er dog kun virksom i de år, hvor sygdommen er under udvikling, og kan derfor ikke anvendes til helt små børn og til ældre. Behandlingen kan ikke kurere sygdommen, men kan give en forbedring af synsfunktionen. Langtidseffekterne af behandlingen er endnu ukendte.
Der foregår i øjeblikket et intenst forsknings- og udviklingsarbejde rettet mod at udvikle behandling til andre arveformer, der giver retinitis pigmentosa.
Hvordan undgår jeg at få eller forværre retinitis pigmentosa?
Retinitis pigmentosa er arvelig. Der findes ikke nogen forholdsregler, som gør det muligt at undgå sygdommens udvikling.
Hvordan udvikler sygdommen sig?
Retinitis pigmentosa kan forløbe på forskellige måder.
Hos nogle patienter kan synstabet være medfødt eller udvikle sig i barndommen. Andre undgår symptomer helt frem til midt i voksenlivet. Mange har et klassisk mønster, hvor det tidligste symptom er besvær med at færdes i mørke, og at synsfeltet gradvist indsnævres.
Efterhånden som sygdommen udvikler sig, mister man mere og mere af det perifere syn, og tilstanden ender med kikkertsyn. Til sidst mister man også centralsynet.
Det kan være vanskeligt at opdage sygdommen i de tidlige stadier, da den udvikler sig langsomt, og man i starten vil vænne sig til det nedsatte syn om natten. Når man bliver klar over, at man er natteblind, kan antallet af stave og tappe i nethinden være reduceret betydeligt, og disse typer celler kan ikke gendannes.
Selv om man har mistet halvdelen af det perifere syn, vil man kun have lette problemer med at færdes til daglige gøremål. Man får problemer med at læse og svært ved at udføre daglige aktiviteter, når synsstyrken bliver tydeligt nedsat. Målinger af følsomheden af disse lysfølsomme sanseceller er langt mere pålidelige, end den beskrivelse man selv kan give af symptomerne.
Behandling af RPE65 associeret retinitis pigmentosa med genterapi kan forsinke sygdommens udvikling, men effekten af behandlingen ud over nogle få år kendes ikke.
Hvordan er langtidsudsigterne?
Sygdommen kan starte i alle aldre. Det er meget forskelligt, hvor opmærksomme folk er på, om de får synstab. Derfor er den alder, hvor man oplever de første symptomer et upræcist mål for, hvor meget sygdommen har udviklet sig. Det giver ikke noget fingerpeg om, hvornår ødelæggelsen af fotoreceptorerne er begyndt.
Generelt er retinitis pigmentosa en sygdom, der udvikler sig med tiden. Man anslår, at synsfeltet aftager med mellem 3 % og 13 % hvert år. Synsstyrken kan forblive god i mange år, fordi der vedbliver at være små områder af fungerende væv i nethindens centrale områder, hvor synsstyrken er størst - selv om sygdommen er langt fremskreden.
På lang sigt er prognosen for synet dårlig. Der er dog forskel fra person til person på, hvor tidligt man kan registrere nedsat syn.
Kørekort
Man vil i de fleste tilfælde ikke opfylde synskravene til kørekort for så vidt angår synsfelt, men vil heller ikke have tilstrækkeligt nattesyn til at kunne køre i lygtetændingstiden
Hvor hyppig er retinitis pigmentosa?
Sygdommen forekommer hos 1 ud af 4000 personer
I Danmark er der cirka 1500 personer med retinitis pigmentosa, mens der på verdensplan er der mere end 1 million mennesker med sygdommen
Retinitis pigmentosa er den hyppigste årsag til blindhed hos personer i alderen 20-64 år i Danmark. Synsnedsættelsen kan begynde i børne- og ungdomsårene eller senere i livet