Epidermolysis bullosa (EB) er en fællesbetegnelse for en gruppe sjældne, arvelige sygdomme, hvor mekaniske påvirkninger af huden fører til blæredannelse på hud og evt. slimhinder
Det latinske navn for blære, eller vabel i huden er bulla
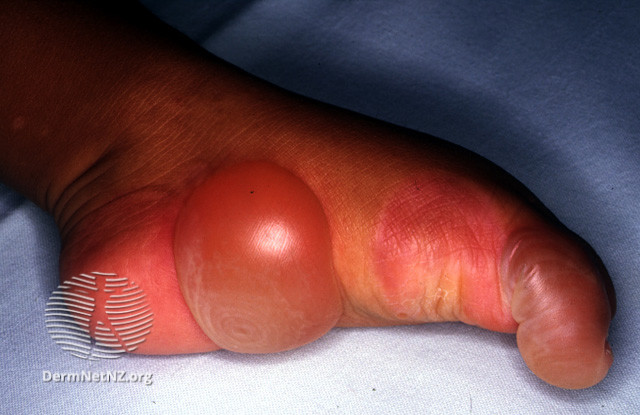
Vablerne brister og efterlader overfladiske, væskende sår
Der er fire hovedgrupper og mange undergrupper
Ved nogle af disse kan komplikationer fra hud og andre organer være invaliderende og livstruende
Der kan opstå sår og komplikationer i øjne og spiserør
EB skyldes defekter i de proteiner, der forankrer overhuden til underhuden
Tidlig diagnose og behandling i et multidisciplinært, højt specialiseret team er afgørende for prognosen ved EB
Basisoplysninger
Synonymer
EB
DEB (dystrofisk EB), JEB (Junctional EB), EBS (EB simplex)
Disse undergrupper blev tidligere kaldt bl.a. Dowling-Meara, Købner, Weber-Cockayne, Herlitz, Hallopeau-Siemens1
Hudens opbygning
Hud og slimhinder har til formål at beskytte kroppen mod udefra kommende påvirkninger som tryk, ændringer i temperatur samt infektioner, og huden medvirker til at regulere kroppens temperatur og væskebalance. Huden er opbygget af tre lag:
Overhud (epidermis), der også består af tre lag
Hornlaget
Vækstlaget
Basalmembranen. Overgangen mellem basalmembran og læderhud kaldes junction
Læderhud (dermis)
Underhud (subcutis)
Hudens funktion er betinget af at cellerne i overhuden sidder fast til hinanden, og at overhuden sidder fast til læderhuden. Den struktur, der holder lagene sammen, er basalmembranen. Det er basalmembranen, som er defekt ved EB.
EB bliver inddelt i fire hovedgrupper alt efter hvilke proteinstrukturer, der er defekte, og dermed afgør hvor blærerne opstår i hud og slimhinder. Alle defekterne er monogenetiske, dvs. at de skyldes mutation i et enkelt gen
Epidermolysis bullosa simplex
Vablerne opstår øverst i basalmembranen
Sår heler ofte uden ardannelse
Bedres ofte i løbet af puberteten
Der findes dog sjældne alvorlige former med involvering af slimhinder og høj dødelighed
Epidermolysis bullosa junctionalis
Vablerne opstår i junction, som er overgangszonen mellem basalmembran og læderhud
Næsten alle har vabler i mundslimhinden
Vablerne opstår hele livet
Der findes dog mildere former med begrænset omfang
Infektion i sårene fører til ardannelse
Epidermolysis bullosa dystrofica
Vablerne opstår nederst i basalmembranen mod læderhuden samt i slimhinder
Sårhelingen fungerer dårligt, og der dannes ar
Kindler epidermolysis bullosa
Vablerne opstår på hænder og fødder
Ofte blæredannelse, også i de indre organer, især i urinveje
Forekomst
Hyppigheden er skønnet til 1 per 50.000 levendefødte1, svarende til ét barn om året i Danmark
Defekter (mutationer) i mindst 18 gener med roller i cellernes evne til at sidde fast til hinanden og til underlaget (adhesion contacts, desmosomer, hemidesmosomer) i hud og slimhinder. De pågældende gener har også betydning for funktionen af andre organer, herunder øjne, mavetarmkanal, luftveje, urinveje og muskler
En del af patienterne har nyopståede mutationer og har derfor ikke arvet sygdommen
Disponerende faktorer
Forekomst i familien (ved autosomal dominante former)
Beslægtede forældre (konsangvinitet), ved autosomal recessive former
Sygdomsbilledet er meget forskelligt alt efter om vablerne opstår over eller under basalmembranen. De alvorlige former kan ses allerede fra fødslen, hvor barnet har blærer flere steder på kroppen. Blærerne opstår i huden ved mekanisk påvirkning med efterfølgende sår, skorper, blødning, ardannelse med skrumpning, infektion, pigmentforandringer, negleforandringer og hårtab
Ved lokaliseret EBS er der kun vabler på hænder, fødder og i mundslimhinde
Ved generaliseret intermediær EBS heler vabler med substanstab (atrofi) og pigmentforandringer. Kan starte ved fødslen eller i tidlige barndom
Ved generaliseret svær EBS er der fra fødslen udbredte forandringer af hud og slimhinder med pigmentforandringer og evt. tab af negle. Bedres med alderen, men forandringer i andre organer end huden giver øget dødelighed
Junctional epidermolysis bullosa (JEB)
Følger som regel autosomal recessiv arvegang
Ved lokaliseret JEB er der vabler på hænder, fødder, albuer og knæ
Ved generaliseret intermediær JEB kommer der mere udbredte vabler med ardannelse, hårtab (alopeci) og forandringer af negle og tændernes emalje. Der kan være forsnævringer (stenoser, strikturer) i luftveje, spiserør og urinveje. Øget risiko for hudkræft (planocellulære karcinomer)
Ved generaliseret svær JEB er der forandringer af huden fra fødslen og komplikationer fra slimhinder i øjne, luftveje, mave- og tarmkanal samt urinveje. Næsten alle dør i første leveår på grund af blodforgiftning (sepsis), lungebetændelse eller obstruktion af luftvejene
Dystrofisk epidermolysis bullosa (DEB)
Følger autosomal dominant eller autosomal recessiv arvegang
Det defekte protein er kollagen type VII (COL7)
Nogle genfejl giver svære forløb med meget sårbar hud, udbredt dannelse af ar med nedsat bevægelighed af led, helt eller delvist tab (amputationer) af fingre/tæer og sammenvoksning (pseudosyndaktyli) af fingre
Ved generaliseret dominant DEB er der som regel kun symptomer fra huden
Ved generaliseret svær recessiv DEB er der forandringer af hud og slimhinder i øjne, mund, mave- og tarmkanal med ardannelse, dårlig ernæring og høj risiko for aggressiv hudkræft, der er den hyppigste dødsårsag
Ved generaliseret intermediær DEB er forløbet mildere
Kindler epidermolysis bullosa
Følger autosomal recessiv arvegang
Vabler på hænder og fødder, fortrinsvis i barnealderen
Senere udvikling af lysfølsomhed og karforandringer (poikilodermi) med substanstab (atrofi) af huden
Komplikationer fra slimhinder i mund, øjne, mavetarmkanal og urinveje
Genterapi. I 2023 blev Vyjuvek godkendt i USA til lokalbehandling af DEB6
Eksperimentelle behandlinger omfatter anden genterapi, celleterapi, autolog hudtransplantation, hvis patienten er genetisk mosaik samt proteinterapi3
Kirurgisk fjernelse af hudkræft
Genetisk rådgivning
EB følger autosomal dominant eller autosomal recessiv arvegang. En del af patienterne har nyopståede mutationer
Ved autosomal dominant arvegang er risikoen for at føre sygdommen videre 50 % ved hver graviditet
Ved autosomal recessiv arvegang er forældrenes risiko for at næste barn får sygdommen 25 %
Når mutation(erne) hos den enkelte patient og forældre er kendt, er der mulighed for prænatal diagnostik eller ægsortering (præimplantationsgenetisk testning, PGT) i eventuelle kommende graviditeter
Særlige behov
Opfølgning i multidisciplinært, højt specialiseret team omfattende bl.a. hudlæger, sygeplejersker, diætister, ergoterapeuter, fysioterapeuter, øjenlæger, tandlæger, børnelæger, ortopæder, mave- og tarmlæger, psykologer, socialrådgivere
Der er som regel et omfattende behov for tværfaglig koordination på tværs af sundheds-, social- og undervisningssektorerne
Indsatser efter behov til barn, familie og netværk for at støtte barnets sociale deltagelse og udvikling
Hjælpemidler efter behov
Ressourcer
VISO - Den nationale videns- og specialrådgivningsorganisation på det sociale område og på specialundervisningsområdet. VISO kan for eksempel rådgive om, hvordan kommunen kan tilrettelægge en faglig indsats, eller hvad et tilbud kan indeholde. VISO yder rådgivning til kommuner, borgere og kommunale, regionale og private tilbud
DUKH - Den Uvildige Konsulentordning på Handicapområdet er en selvejende institution under Social- og Indenrigsministeriet. Medvirker til at styrke retssikkerheden for mennesker med et handicap ved at give uvildig rådgivning til mennesker med handicap og deres pårørende i sager, der måske er gået i hårdknude, eller hvor borgeren føler sig uretfærdigt behandlet
Sjældne Diagnoser - Selvstændig paraplyorganisation for små, landsdækkende patientforeninger for familier og voksne med sjældne sygdomme og handicap, se www.sjaeldnediagnoser.dk. Sjældne Diagnoser har en Helpline/telefonrådgivning, som tilbyder information, rådgivning og mestringsstøtte, og som kan hjælpe med kontakt til andre patienter og pårørende
Sjældne-netværket - Sjældne-netværket er et tilbud til mennesker med sjældne sygdomme og handicap samt til deres pårørende, der ikke har en forening i Danmark at henvende sig til og måske melde sig ind i. Sjældne-netværket administreres af Sjældne Diagnoser, der er paraplyorganisation for hovedparten af foreningerne på sjældneområdet. Sjældne-netværkets formål er at skabe kontakt mellem personer og familier, der lever med den samme sjældne sygdom. Sjældne Diagnoser har et netværk for epidermolysis bullosa simplex
Placering i sundhedsvæsenet
Ifølge Sundhedsstyrelsens specialevejledning for dermatologi 2023 er hudafdelingerne på Herlev og Gentofte Hospital, Odense Universitetshospital og Aarhus Universitetshospital godkendt til at varetage behandlingen af epidermolysis bullosa congenita i samarbejde med Centrene for Sjældne Sygdomme, klinisk genetik, pædiatri og andre relevante specialer
Has C et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders of skin fragility. Br J Dermatol. 2020; 183.; 614-27.
PubMed
Lucky AW, Whalen J, Rowe S, Marathe KS, Gorell E. Diagnosis and Care of the Newborn with Epidermolysis Bullosa. Neoreviews. 2021; 22.; e438-e451.
PubMed
Has C, Liu L, Bolling MC, Charlesworth AV, El Hachem M, Escámez MJ, Fuentes I, Büchel S, Hiremagalore R, Pohla-Gubo G, van den Akker PC, Wertheim-Tysarowska K, Zambruno G. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020; 182.; 574-592.
PubMed
Khan A, Riaz R, Ashraf S, Akilimali A. Revolutionary breakthrough: FDA approves Vyjuvek, the first topical gene therapy for dystrophic epidermolysis bullosa. Ann Med Surg (Lond). 2023; 85.; 6298-6301.
PubMed
Fagmedarbejdere
Flemming Skovby
Speciallæge i pædiatri og klinisk genetik, tidl. overlæge, professor, dr.med., Sjællands Universitetshospital
Bemærk venligst, at du IKKE vil modtage svar på henvendelser, der omhandler din egen sygdom, pårørendes sygdom, blodprøvesvar, hjælp til at udarbejde skoleopgaver og litteratursøgning.