Arvelig tilstand karakteriseret ved pigmenteringer på hud og slimhinder, især omkring munden med fregner på læberne, polypper i mave- og tarmkanalen og øget risiko for en række tumorer i og udenfor mave- og tarmkanalen
Livslang overvågning har til formål at forhindre komplikationer fra mave- og tarmkanalen og at opdage ondartede tumorer i et tidligt stadie
Basisoplysninger
Synonymer
PJS
Hamartomatøs intestinal polypose
Forekomst
Ukendt; skøn varierer fra 1 per 25.000 til 1 per 200.000
Fejl i genet for serin/threonin proteinkinase, STK11, der koder for en tumor suppressor, i 70-90 % af familier med PJS
Fejlen kan være en enkelt basevariant (mutation) i genet eller en deletion, hvor der mangler en del af eller hele genet
Disponerende faktorer
Ingen kendte
Sygdomstegn
Peutz-Jeghers-polypper kan forekomme overalt i mave- og tarmkanalen, men hyppigst i tyndtarmen. Polypperne bliver kaldt hamartomer ud fra deres karakteristiske udseende ved mikroskopisk undersøgelse, og de er godartede.2 De kan også forekomme andre steder, f.eks. i næse eller galdeblære.
Inden 20-års alderen har 50 % af patienter haft symptomer fra deres polypper. Antallet af polypper er meget forskelligt fra person til person: nogle har meget få, mens andre i løbet af deres levetid udvikler over 100 polypper.
PJS debuterer oftest i løbet af de første 10 leveår med
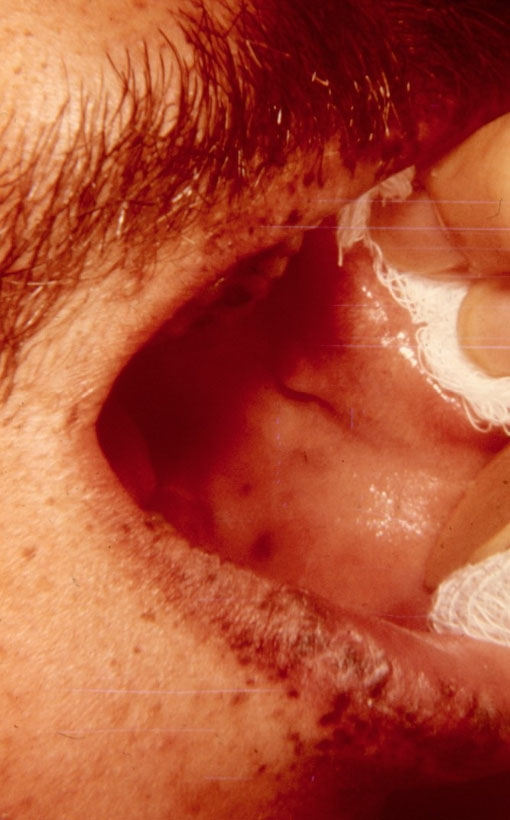
Mukokutane pigmenteringer, dvs. mørkeblå-mørkebrune pletter (maculæ) i huden omkring mund (se foto), øjne og næsebor, i mundslimhinde, omkring endetarmsåbning, og på fingre og tæer
Akut blokering af tyndtarmen på grund af polypper
Blødning fra polypper
Blodmangel (anæmi) på grund af blødning fra polypper
I voksenalderen er forløbet præget af tumorer i og udenfor mave- og tarmkanalen. Den ophobede (kumulative) risiko for en ondartet tumor stiger fra 5 % til 31 % til 85 % i henholdsvis 30-, 50- og 70-årsalderen. Der er tale om tumorer i flere forskellige organer, men hyppigst i mave- og tarmkanalen (mavesæk, tyndtarm, tyktarm), bryster, underliv (æggestokke og livmoderhals) og bugspytkirtlen.
De ondartede tumorer udvikler sig oftest i voksenalderen.
Diagnose
Baseret på de kliniske fund hos en patient med 1) to eller flere PJ polypper bekræftet ved biopsi, 2) PJ polyp(per) og familieanamnese for PJS, 3) karakteristiske mukokutane pigmenteringer og en familieanamnese for PJS, eller 4) PJ polyp(per) og karakteristiske mukokutane pigmenteringer
Bekræftes ved fund af fejl i STK11-genet (blodprøve)
Overvågning (screening) er en del af behandlingen ved PJS, og den har til formål at
Finde og fjerne polypper i tarmen som kunne give anledning til komplikationer, f.eks. obstruktion eller blødning
Finde ondartede tumorer i et tidligt stadie
Overvågningsprogrammet følger de danske retningslinjer på området og bliver løbende tilpasset den nyeste viden
Drenge bliver fulgt fra første leveår med årlige testikelundersøgelser og begge køn fra 8-års alderen med regelmæssige kikkertundersøgelser/kapselundersøgelser af tarmen
Voksne bliver tilbudt yderligere undersøgelser af fx bryster og underliv
Selve behandlingen af opståede mave- og tarmkomplikationer og fundne tumorer følger gældende retningslinjer
Genetisk rådgivning
PJS følger autosomal dominant arvegang. Det vil sige at en patient skal have patogen variant (mutation) i et af sine to STK11-gener, enten i det fra moderen eller i det fra faderen, for at få sygdommen
Omkring halvdelen af patienter har PJS på baggrund af en nyopstået genfejl (nymutation)
Der er fuld penetrans. Det vil sige at alle med en patogen variant i STK11 vil få symptomer på et eller andet tidspunkt i livet
Der er varierende ekspressivitet. Det vil sige at på trods af samme mutation er der variation mellem forskellige personer og familier
En far eller mor med sygdommen har 50 % risiko for at videreføre den patogene variant og dermed PJS til hvert barn
Hvis familiens patogene variant er kendt, er der mulighed for foster (prænatal) diagnostik ved moderkagebiopsi i første trimester og evt. for ægsortering
VISO - Den nationale videns- og specialrådgivningsorganisation på det sociale område og på specialundervisningsområdet. VISO kan for eksempel rådgive om, hvordan kommunen kan tilrettelægge en faglig indsats, eller hvad et tilbud kan indeholde. VISO yder rådgivning til kommuner, borgere og kommunale, regionale og private tilbud
DUKH - Den Uvildige Konsulentordning på Handicapområdet er en selvejende institution under Social- og Indenrigsministeriet. Medvirker til at styrke retssikkerheden for mennesker med et handicap ved at give uvildig rådgivning til mennesker med handicap og deres pårørende i sager, der måske er gået i hårdknude, eller hvor borgeren føler sig uretfærdigt behandlet
Sjældne Diagnoser - Selvstændig paraplyorganisation for små, landsdækkende patientforeninger for familier og voksne med sjældne sygdomme og handicap, se www.sjaeldnediagnoser.dk. Sjældne Diagnoser har en Helpline/telefonrådgivning, som tilbyder information, rådgivning og mestringsstøtte, og som kan hjælpe med kontakt til andre patienter og pårørende
Sjældne-netværket - Sjældne-netværket er et tilbud til mennesker med sjældne sygdomme og handicap samt til deres pårørende, der ikke har en forening i Danmark at henvende sig til og måske melde sig ind i. Sjældne-netværket administreres af Sjældne Diagnoser, der er paraplyorganisation for hovedparten af foreningerne på sjældneområdet. Sjældne-netværkets formål er at skabe kontakt mellem personer og familier, der lever med den samme sjældne sygdom. Sjældne Diagnoser har et netværk for PJS
Placering i sundhedsvæsenet
Børnekirurgiske afdelinger og de regionale mavetarmkirurgiske afdelinger i samarbejde med klinisk genetik; genetisk rådgivning om PJS er en højtspecialiseret funktion
Jelsig AM, Qvist N, Sunde L, Brusgaard K, Hansen T, Wikman FP, Nielsen CB, Nielsen IK, Gerdes AM, Bojesen A, Ousager LB. Disease pattern in Danish patients with Peutz-Jeghers syndrome. Int J Colorectal Dis. 2016; 31.; 997-1004.
PubMed
Jelsig AM. Hamartomatous polyps - a clinical and molecular genetic study. Dan Med J. 2016; 63..
PubMed
Wagner A, Aretz S, Auranen A, Bruno MJ, Cavestro GM, Crosbie EJ, Goverde A, Jelsig AM, Latchford A, Leerdam MEV, Lepisto A, Puzzono M, Winship I, Zuber V, Möslein G. The Management of Peutz-Jeghers Syndrome: European Hereditary Tumour Group (EHTG) Guideline. J Clin Med. 2021; 10..
PubMed
Fagmedarbejdere
Flemming Skovby
Speciallæge i pædiatri og klinisk genetik, tidl. overlæge, professor, dr.med., Sjællands Universitetshospital
Anne Marie Jelsig
Afdelingslæge, Afdeling for Genetik, Rigshospitalet
Erling Peter Larsen
Speciallæge i almen medicin, Silkeborg
Har du en kommentar til artiklen?
Bemærk venligst, at du IKKE vil modtage svar på henvendelser, der omhandler din egen sygdom, pårørendes sygdom, blodprøvesvar, hjælp til at udarbejde skoleopgaver og litteratursøgning.