Porfyria cutanea tarda (PCT) er en samlebetegnelse for en gruppe af både erhvervede og arvelige tilstande, hvor der ses en mangel på aktivitet af enzymet ansvarlig for hæm-syntesen, nemlig uroporphyrinogen decarboxylase (UROD)
PCT er den hyppigste porfyri og udgør 80-90 % af tilfældene i almen praksis
Pga. den nedsatte enzymaktivitet ses en øget dannelse af fotoaktive porfyriner, der absorberer energi i det synlige violette lysspektrum. Dette medfører oxidative skader i vævet, synligt i form af bulløse hudlæsioner
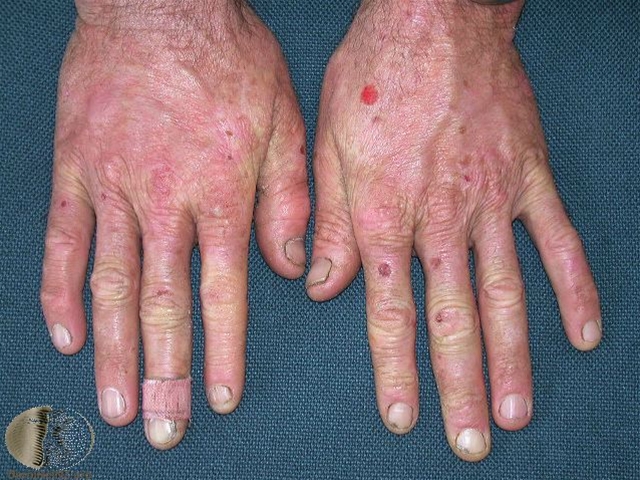
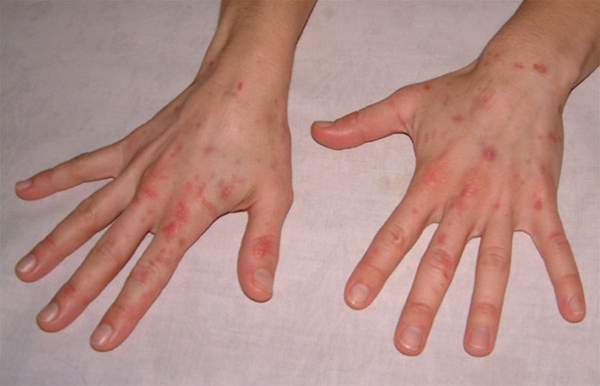
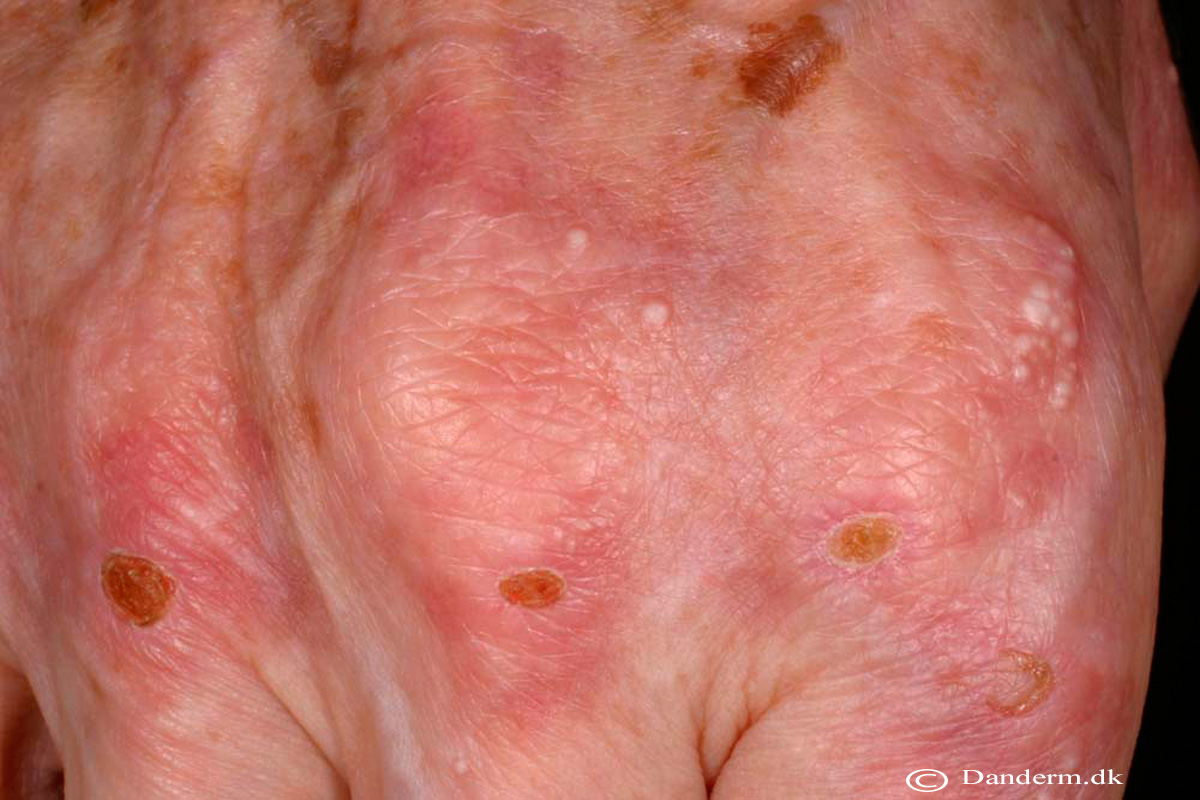
Skrøbelig hud med erosioner, sårdannelse og millier ofte på håndryggene
Påvisning af øget urinudskillelse af porfyriner
Evt. stansebiopsi fra huden inkl. immunoflourescens undersøgelse, mest for at udelukke andre bulløse hudsygdomme
Sygehistorie
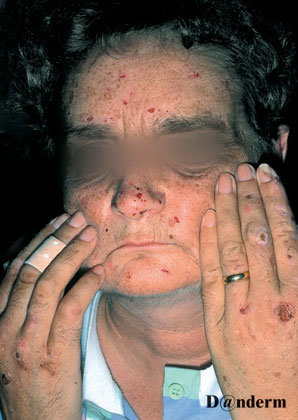
Rødme, blærer, sår i huden opstår efter soleksponering - også igennem glasruder
Sporadisk, erhvervet PCT manifesterer sig i voksenalderen
Ved den familiære form er reporteret symptomer hos heterozygote allerede i barnealderen, og tidlige symptomer ses ofte hos de homozygote
Der ses udbrud af bullae eller vesikler på lyseksponeret hud
Urinen ændrer farve - farven ofte som te eller vin
Kliniske fund
Vesikler og bullae, oftest på håndrygge, som kan blive flere centimeter store
Bullae kan i sjældne tilfælde være hæmorrhagiske, og let smertefulde
Milie dannelse i huden især på håndrygge
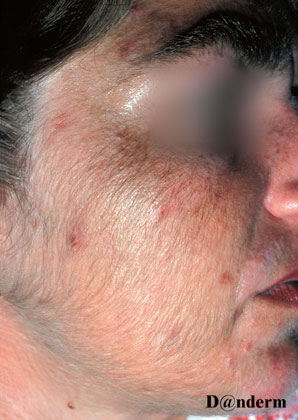
Evt. hyperpigmenteringer, hypotrofiske ar eller pletvis sclerodermisk hud
Der er ofte øget behåring og aktiniske forandringer i huden
Ved svære tilfælde kan ses cikatricielt hårtab og negleforandringer
Urin fluorescerer pink i Woods light
Supplerende undersøgelser i almen praksis
Urinprøve til porfyri-undersøgelse
Patienter med PCT har et specifikt mønster med øget indhold af uro-, hepta-, and hexaporfyrin. Urinprøven skal beskyttes med lys (pakkes i sølvpapir el. lign.)
Andre undersøgelser hos specialist eller undersøgelse på sygehus
Ved mistanke om anden bulløs hudsygdom tages ofte en hudbiopsi til histologi og immunoflourescens undersøgelse
Ca. 80 % af patienterne har den erhvervede form, 20 % den arvelige form, men der kan være geografiske variationer
Derfor evt. blodprøve til UROD-måling ved mistanke om den arvelige form
Familiær PCT skyldes ofte en autosomal dominant arvegang med en enkelt mutation i UROD genet, der er dog beskrevet 121 forskellige mutationer i Human Genome Mutation Database
De fleste patienter udredes også med gentest for hæmokromatose, HFE-genet
Erhvervet PCT i befolkningsgrupper udsat for polyhalogenerede aromatiske hydrocarbon hepatotoxiner er beskrevet, og kaldt for "epidemisk” porphyria cutanea tarda
Ultralydsscanning af leveren kan være relevant
Differentialdiagnoser
Andre bulløse hudlidelser, fx pemfigoid og pemphigus
Diabetiske bullae
Erythropoietisk protoporfyri
Hos børn: kongenit epidermolysis bullosa
Erhvervet epidermolysis bullosa
Pseudoporfyri (fx ved svær nyresygdom)
Andre fotodermatoser, f.eks. phytofotodermatoser efter udsættelse for bjørneklo eller visse citrusfrugter
Bulløs kutan lupus erythematosus
Behandling
Behandlingsmål
Holde patienterne symptomfrie
Opdage udvikling af leversygdom
Generelt om behandlingen
Det er som regel adækvat behandling at tømme jerndepoterne ved venesectio
450 ml pr. uge indtil patienten er klinisk bedre, eller Hb under 6,5-7,0 mmol/L og ferritin under 15-22 µg/L, dvs laveste værdi i det alders- og kønsbetingede referenceinterval
Ved klinisk sygdom er der anbefalet forsigtighed med potentielt hepatotoksiske medikamenter, som bliver metaboliseret i cytokrom P450-systemet
Der suppleres med Hb, leuko + diff, levertal og hvis relevant screening for hepatitis B og C
Ved mistanke om leversygdom henvises til ultralyd af lever eller fibroscanning (en form for ultralydsundersøgelse til at vurdere bindevævsudvikling/fibrose leveren)
Hvis kvinder med PCT I rolig fase har behov for østrogen substitution, kan det forsøges med transdermale plastre
Råd til patienten
Beskyt dine hænder ved manuelt arbejde, hold eventuelle blærer og sår rene, anvend skumplastre, anvend fugtighedscreme dagligt
Anvend solbeskyttelse, med solcreme med fysisk filter og beklædning for at undgå at udvikle blærer
Anvend solcremer med titanium dioxid eller zinc oxid, da kun solcreme med fysiske filtre har effekt
Rygestop
Stop alkoholforbrug
Mød op til de halvårlige leverfunktions kontroller
Medicinsk behandling
Behandling med hydroxychloroquin i en lille dosering 100-200 mg x 2 ugentligt er en mulighed, men skal foregå i samarbejde med en hudlæge og evt. en gastroenterolog/hepatolog og ved nyresygdom i samråd med nefrolog. Ikke indiceret ved kronisk anæmi.
For anæmiske PCT-patienter, er human rekombinant erythropoietin en behandlingsmulighed i hospitalsregi
Desferal, deferoxamin en jern-chelator kan anvendes i særlige tilfælde
Forebyggende behandling
Undgå østrogenbrug eller overdosering af jern
Nedsæt eller stop indtag af alkohol
Begræns indtag af rødt kød
Rygestop
Indtag sufficient mængde C-vitamin
Henvisning
Henvises til dermatologisk afdeling, evt. direkte til klinisk genetisk afdeling samt medicinsk afdeling afhængig af symptomatologi
Det anbefales, at patienterne bliver udredt i samarbejde med en hudlæge, og ved tegn på leverlidelse, en gastroenterolog el. hepatolog. En nefrolog vil ofte allerede være involveret ved svær nyresygdom
Opfølgning
Plan
Kontrol ca. hvert halve år i starten er anbefalet
U-porfyriner evt. årligt afhængigt af symptomer
Patienterne kan i stabil fase evt. følges ved egen læge eller i dermatologisk speciallægepraksis.
Hvad bør man kontrollere
Hos personer over 55 år anbefaler man hvert eller hvert andet år:
Måle urin porfyrin-udskillelsen
Kontrol af jerndepot og leverenzymer
Ultralyd af leveren hvert 2. år
Sygdomsforløb, komplikationer og prognose
Sygdomsforløb
Ofte starter symptomerne på PCT med skrøbelig hud og blære- eller sårdannelse på håndryggene
De mange blærer i huden øger risikoen for infektion i huden og kan hæmme patientens arbejdsevne i perioder
Komplikationer
God, hvis tilstanden behandles. Tidligere blev rapporteret en øget risiko for leverkræft, men det mener man nu skyldes hepatitis, og ikke PCT i sig selv
Prognose
God, hvis der ikke er kompliceret leversygdom
Langt de fleste opnår kontrol af hudsymptomer ved at eliminere provokerende faktorer og fortsat venesectio efter behov
Baggrundsoplysninger
Definition
Porfyria cutanea tarda (PCT) er en bulløs hudsygdom forårsaget af ophobning af porfyrin-metabolitter på grund af arvelige eller erhvervede enzymmangler i leverens hæm-syntese1,2
PCT er en samlebetegnelse for en gruppe af både erhvervede (spontane og intermitterende) og arvelige tilstande, hvor der ses en mangel på aktivitet af enzymet ansvarlig for hæm-syntesen, nemlig uroporphyrinogen decarboxylase (UROD)
Pga den nedsatte enzymaktivitet ses en øget dannelse af abnorme fotoaktive porfyriner, der er molekyler, som absorberer energi i det synlige violette lysspektrum. Dette medfører oxidative skader i vævet, synligt i form af bullæse hudlæsioner
Reduceret UROD enzyme aktivitet medfører ophobning af hæm-syntese metabolitter i hepatocytter. En jern-medieret spontan oxidering til phototoksiske porfyriner opstår. Disse fotoaktive porfyriner forlader leveren og akkumuleres i huden, og kan også identificeres i blod, urin og fæces
Der kan påvises en markant øget udskillelse af porfyriner i urinen
Prævalens af PCT er ca. 1:25.000-100.000 indbyggere
Ca. 80 % af alle PCT-tilfælde er erhvervede; 20 % er genetiske, men ratioen varierer mellem etniske grupper og i forskellige verdensdele.
Lige hyppigt hos mænd og kvinder
PCT udgør 90 % af alle porfyrier
De øvrige porfyriformer er langt mere sjældne
Geografi/klima/etnicitet
Højere prævalens af PCT ses i Europæiske populationer. Men også høj prævalens hos Bantu befolkningen i Sydafrika, formentlig pga hepatisk siderosis. Den familiære PCT varierer også på verdensplan, og udgør hhv 14.6 % i Spanien, 20-24 % i Danmark og 50 % i Chile.
Ætiologi og patogenese
PCT skyldes mangelfuld funktion af leverenzymet uroporphyrinogen decarboksylase (UROD). Dette fører til ophobning af fototoksiske porfyriner i huden
Den genetiske form for porphyria cutanea tarda skyldes ofte autosomal dominant arvegang med en single mutation i UROD -genet
Betydelig variation i fænotypisk penetrans
De fleste med mutationen og nedsat enzymaktivitet vil som regel ikke få symptomer (latent sygdom), men kan få sygdom, hvis de bliver udsat for udløsende faktorer (se næste afsnit)
Den mere almindelige erhvervede form forekommer hos individer, der har normale UROD DNA-sekvenser, men har en ændret levermetabolisme med særlig tilbøjelighed til at hæmme UROD aktiviteten
Disponerende faktorer
Udløsende faktorer:
Alkoholoverforbrug
Østrogener
For høj indtagelse af jern
Nogle miljøgifte, dette skal mistænkes ved pludselig ophobning af PCT i geografiske regioner
Bemærk venligst, at du IKKE vil modtage svar på henvendelser, der omhandler din egen sygdom, pårørendes sygdom, blodprøvesvar, hjælp til at udarbejde skoleopgaver og litteratursøgning.