Ved mistanke om syndromal kraniosynostose foretages molekylærgenetiske undersøgelser
Behandling
Operation i 3-9-mdrs.alderen
Henvisning
Ved mistanke om kraniesynostose henvises til lokal børneafdeling, som tager stilling til viderehenvisning til det kraniofaciale team på Rigshospitalet eller Aarhus Universitet Hospital
Diagnose
Diagnostiske kriterier
Diagnosen baserer sig på kliniske fund, CT og molekylærgenetiske undersøgelser
Sygehistorie
Kraniosynostose er oftest til stede ved fødslen, men tilstanden bliver ikke altid diagnosticeret, hvis den er mild
Som regel diagnosticeres en kraniedeformitet i løbet af de første måneder af livet
Afklar følgende
Hvilken stilling sover barnet i?
For at skelne kraniosynostose fra plagiocefali uden synostose
Kliniske fund
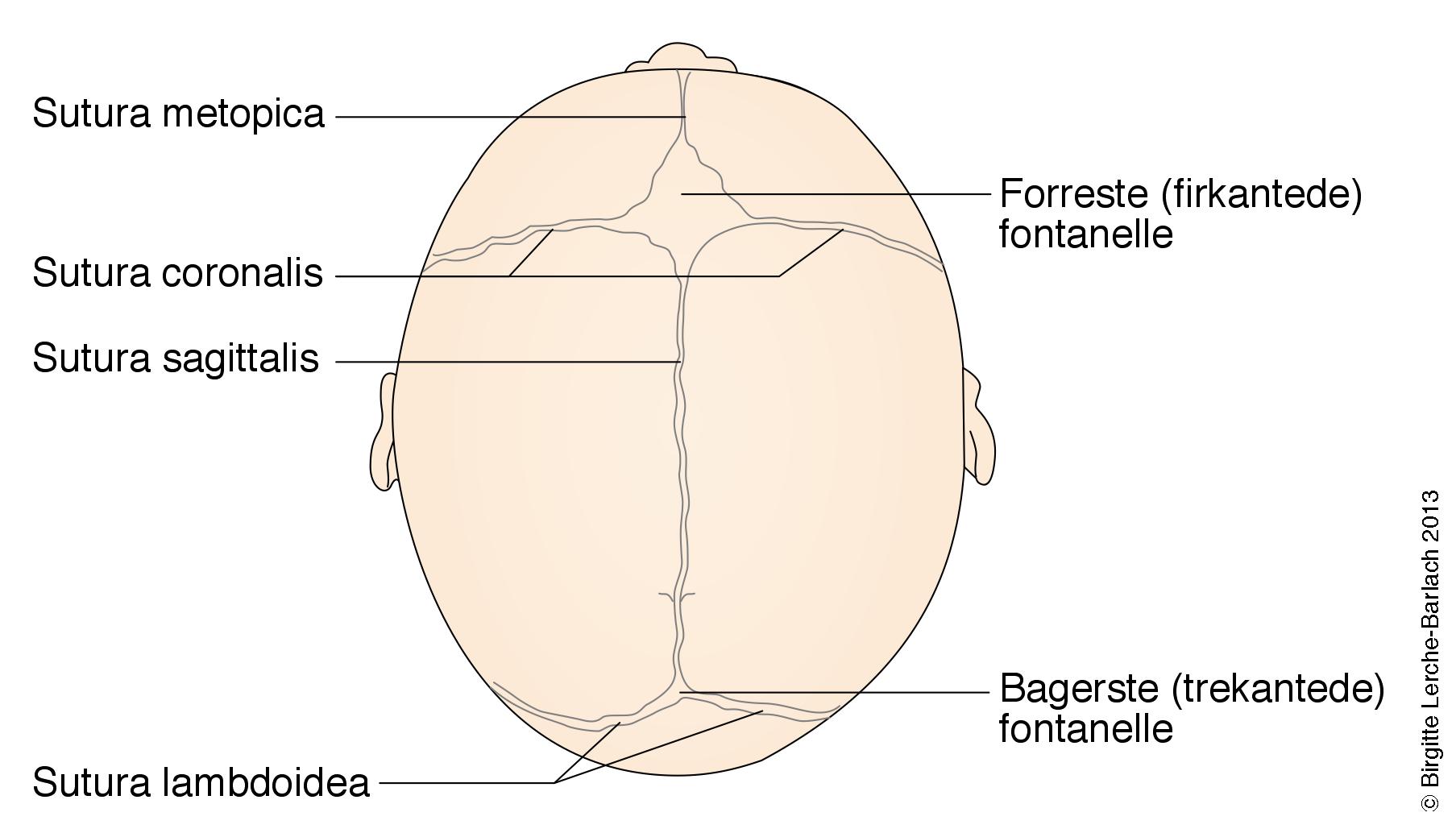
Formen på kalvariet er typisk for hver af de forskellige typer sutursynostoser
Målinger af hovedomfanget er afgørende for at påvise ledsagende mikrocefali eller makrocefali (forårsaget af hydrocefalus)
Børn med kraniofaciale misdannelser bør gennemgå en grundig undersøgelse af nakke, ryg, fingre og tæer
Supplerende undersøgelser i almen praksis
Måling af hovedomfang og palpation af kranium mhp. vurdering af fontaneller
Andre undersøgelser hos specialist
Molekylærgenetisk undersøgelse ved mistanke om syndrom1
Klinisk undersøgelse er som regel tilstrækkelig til at diagnosticere enkelt-sutur kraniosynostose,
CT giver et tydeligt billede af suturerne
Herudover bidrager CT til vurderingen af evt. strukturelle ændringer i hjernen (f.eks. hydrocefalus, agenese af corpus callosum) og til at ekskludere andre årsager til asymmetrisk vækst (f.eks. hjernehemiatrofi, kronisk subduralt hæmatom)
Tredimensionel rekonstruktion af CT-billeder er en hjælp for kirurgen. som planlægger rekonstruktion af kraniofacial deformitet
Håndteringen af et barn med kraniofaciale misdannelser kræver tværfagligt teamarbejde
Barnet skal helst vurderes i løbet af de første uger af livet
Behandlingen er kirurgisk korrektion
Håndtering i almen praksis
Ved mistanke henvises elektivt til børneafdeling. Ved tegn på øget intrakranielt tryk akut
Råd til patienten
Ingen
Kirurgi
Bedste tidspunkt for behandling er i 3-9-mdrs.alderen
Ved øget intrakranielt tryk er hurtig dekompression påkrævet
Kirurgisk intervention ved ikke-syndromal kraniosynostose
Sagittal synostose
Indgrebet indebærer enten en stribeformet kraniektomi eller remodellering af kraniet med fjernelse af involverede knogler, som så trimmes, reformes og fikseres
Der er rapporteret succes med anvendelse af en minimalt invasiv endoskopisk metode4
Coronal synostose
Formålet er at øge den anteroposteriore længde af kalvariet
Metopisk synostose
Formålet er at øge volumet af forreste kraniale fossa
Kirurgisk intervention ved syndromal kraniosynostose5,3
Indebærer korrektion af kraniosynostosen i 3-6-mdrs.alder, og evt. korrektion af ekstremitetsdefekter i 1-2-årsalder
Som ung voksen udføres indgreb for at normalisere udseende og korrigere malokklusion, forudgået af ofte langvarig ortodontisk behandling
Tæt opfølgning er vigtig efter kirurgi for at kontrollere, at suturerne ikke gror sammen igen
Forebyggende behandling
Ingen
Henvisning
Ved mistanke om kraniosynostose henvises til den lokale børneafdeling, som vil viderehenvise til det kraniofaciale team på Aarhus Universitet Hospital eller Rigshospitalet, såfremt diagnosen bekræftes
De vigtigste komplikationer forbundet med ukorrigeret kraniosynostose er øget intrakranielt tryk, asymmetri af ansigtet, forsinket udvikling, malokklusion og manglende samsyn
Asymmetri af orbitae fører til skelen med risiko for unilateralt synstab
Skyldes mutation i genet for fibroblast vækst faktor receptor 2, FGFR2, oftest nymutation
Autosomal dominant tilstand, som forekommer hos 1 af 160.000 nyfødte
Symmetrisk syndaktyli af alle ekstremiteter
Andre kliniske fund inkluderer misdannet kranium, hypertelorisme, midfacial hypoplasi, choanalatresi- eller stenose og smalle orbitae
Intrakranielle misdannelser forekommer med variabel mental retardering
Kardiale og renale misdannelser kan også forekomme
Disponerende faktorer
Familiær ikke-syndromal kraniosynostose hos 2-6 % af børn med sagittal synostose og 8-14 % af børn med coronal synostose. Overføres som en autosomal dominant tilstand
Wilkie AO, Bochukova EG, Hansen RM, Taylor IB, Rannan-Eliya SV, Byren JC, Wall SA, Ramos L, Venâncio M, Hurst JA, O'rourke AW, Williams LJ, Seller A, Lester T. Clinical dividends from the molecular genetic diagnosis of craniosynostosis. Am J Med Genet A. 2007; 143A.; 1941-9.
PubMed
Blessing M, Gallagher ER. Epidemiology, Genetics, and Pathophysiology of Craniosynostosis. Oral Maxillofac Surg Clin North Am. 2022; 34.; 341-352.
PubMed
Agochukwu NB, Doherty ES, Muenke M. Muenke Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Eds. GeneReviews May 10, 2006.
Vis kilde
Professor, ph.d., praktiserende læge, Institut for Folkesundhed - Almen Medicin, Aarhus Universitet
Har du en kommentar til artiklen?
Bemærk venligst, at du IKKE vil modtage svar på henvendelser, der omhandler din egen sygdom, pårørendes sygdom, blodprøvesvar, hjælp til at udarbejde skoleopgaver og litteratursøgning.