Referencer
- Jamis-Dow CA, Turner J, Biesecker LG, Choyke PL. Radiologic manifestations of Proteus syndrome. Radiographics. 2004; 24.; 1051-68. PubMed
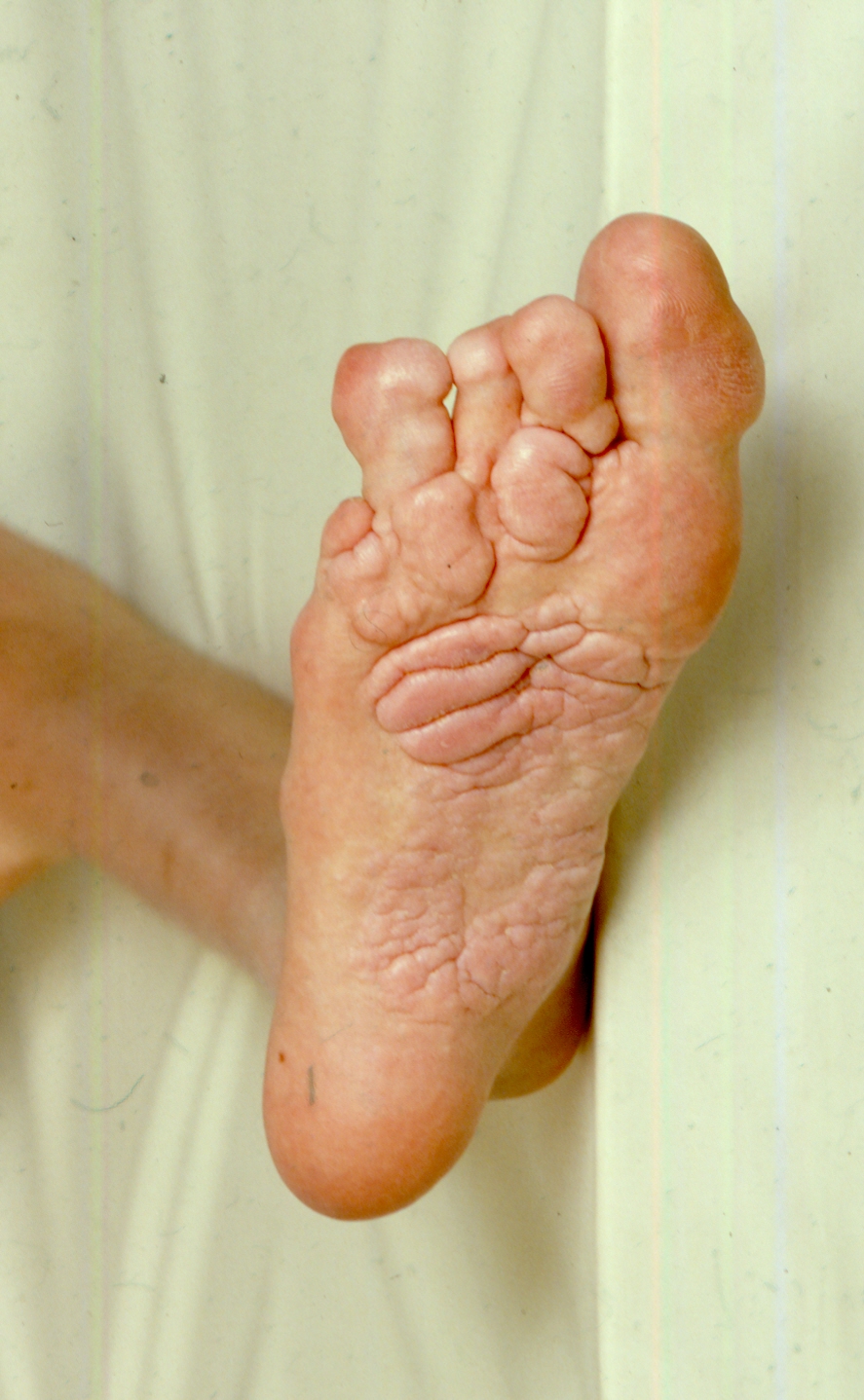
- Pithadia DJ, Cartron AM, Biesecker LG, Darling TN. Dermatologic findings in individuals with genetically confirmed Proteus syndrome. Pediatr Dermatol. 2021; 38.; 794-799. PubMed
- Takyar V, Khattar D, Ling A, Patel R, Sapp JC, Kim SA, Auh S, Biesecker LG, Keppler-Noreuil KM, Heller T. Characterization of the hepatosplenic and portal venous findings in patients with Proteus syndrome. Am J Med Genet A. 2018; 176.; 2677-2684. PubMed
- Keppler-Noreuil KM, Lozier JN, Sapp JC, Biesecker LG. Characterization of thrombosis in patients with Proteus syndrome. Am J Med Genet A. 2017; 173.; 2359-2365. PubMed
- Sapp JC, Hu L, Zhao J, Gruber A, Schwartz B, Ferrari D, Biesecker Md LG. Quantifying survival in patients with Proteus syndrome. Genet Med. 2017; 19.; 1376-1379. PubMed