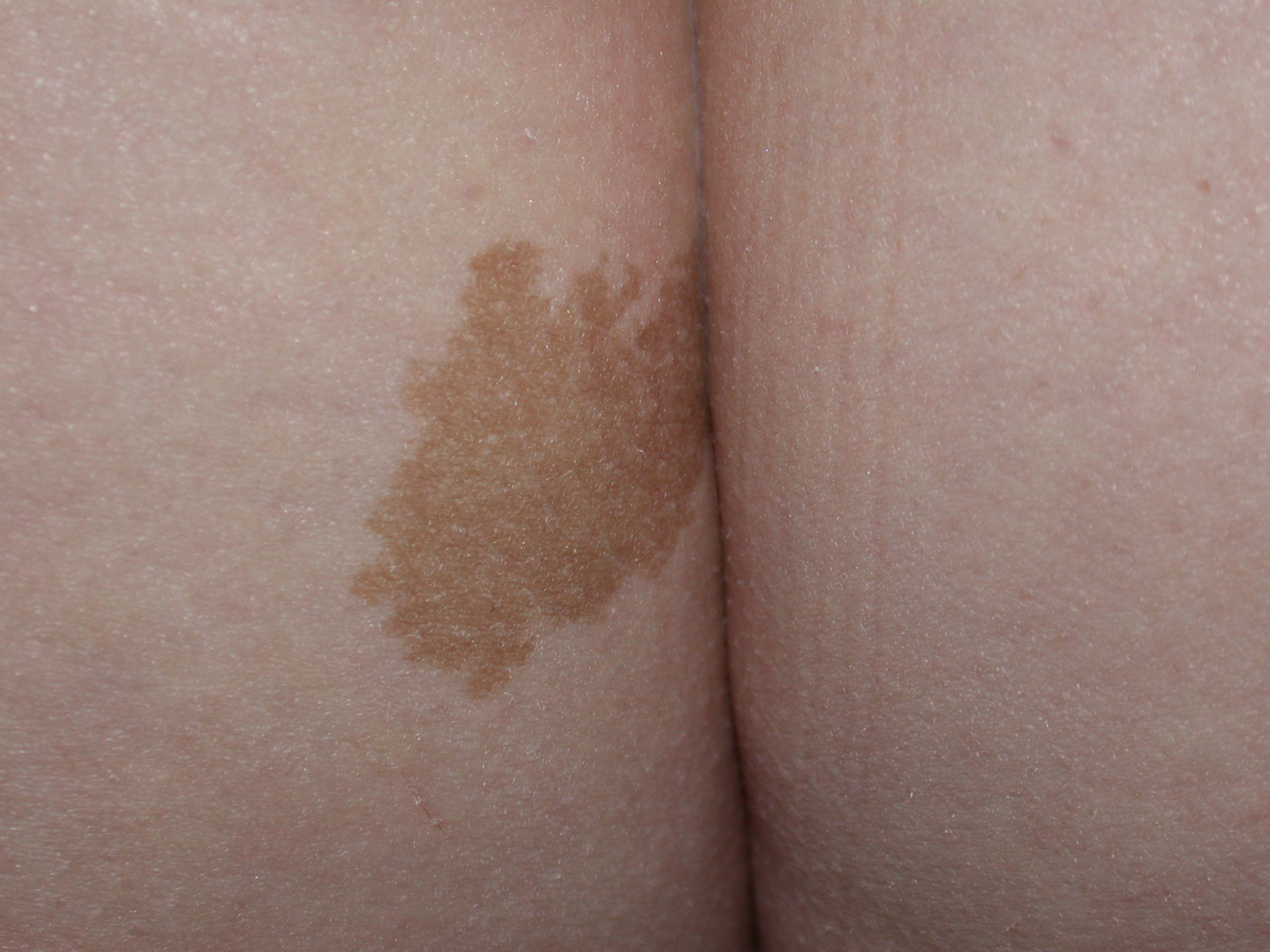
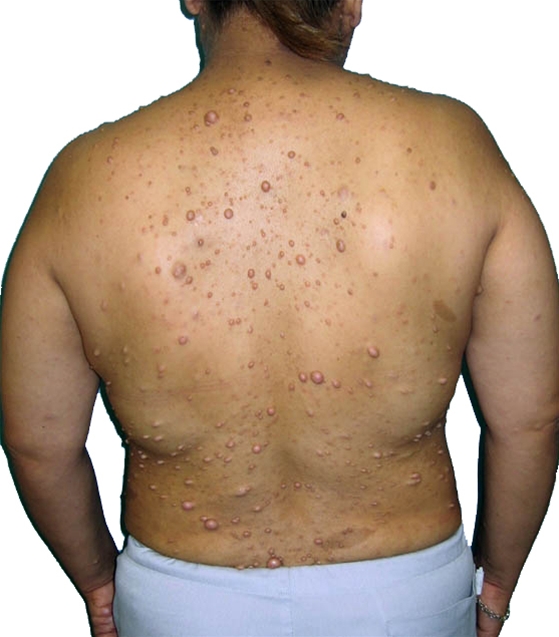
Referencer
- Friedrich RE, Nuding MA. Optic Pathway Glioma and Cerebral Focal Abnormal Signal Intensity in Patients with Neurofibromatosis Type 1: Characteristics, Treatment Choices and Follow-up in 134 Affected Individuals and a Brief Review of the Literature. Anticancer Res. 2016; 36.; 4095-121. PubMed
- Vogel AC, Gutmann DH, Morris SM. Neurodevelopmental disorders in children with neurofibromatosis type 1. Dev Med Child Neurol. 2017; 59.; 1112-1116. PubMed
- Roy A, Roulin JL, Gras-Le Guen C, Corbat ML, Barbarot S. Executive functions and quality of life in children with neurofibromatosis type 1. Orphanet J Rare Dis. 2021; 16.; 420. PubMed
- Lehtonen A, Howie E, Trump D, Huson SM. Behaviour in children with neurofibromatosis type 1: cognition, executive function, attention, emotion, and social competence. Dev Med Child Neurol. 2013; 55.; 111-25. PubMed
- Morris SM, Acosta MT, Garg S, Green J, Huson S, Legius E, North KN, Payne JM, Plasschaert E, Frazier TW, Weiss LA, Zhang Y, Gutmann DH, Constantino JN. Disease Burden and Symptom Structure of Autism in Neurofibromatosis Type 1: A Study of the International NF1-ASD Consortium Team (INFACT). JAMA Psychiatry. 2016; 73.; 1276-1284. PubMed
- Elefteriou F, Kolanczyk M, Schindeler A, Viskochil DH, Hock JM, Schorry EK, Crawford AH, Friedman JM, Little D, Peltonen J, Carey JC, Feldman D, Yu X, Armstrong L, Birch P, Kendler DL, Mundlos S, Yang FC, Agiostratidou G, Hunter-Schaedle K, Stevenson DA. Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet A. 2009; 149A.; 2327-38. PubMed
- Ejerskov C, Krogh K, Ostergaard JR, Fassov JL, Haagerup A. Constipation in adults with neurofibromatosis type 1. Orphanet J Rare Dis. 2017; 12.; 139. PubMed
- Ejerskov C, Krogh K, Ostergaard JR, Joensson I, Haagerup A. Gastrointestinal Symptoms in Children and Adolescents With Neurofibromatosis Type 1. J Pediatr Gastroenterol Nutr. 2018; 66.; 872-875. PubMed
- Stewart DR, Korf BR, Nathanson KL, Stevenson DA, Yohay K. Care of adults with neurofibromatosis type 1: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018; 20.; 671-682. PubMed
- Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017; 3.; 17004. PubMed
- Nguyen R, Jett K, Harris GJ, Cai W, Friedman JM, Mautner VF. Benign whole body tumor volume is a risk factor for malignant peripheral nerve sheath tumors in neurofibromatosis type 1. J Neurooncol. 2014; 116.; 307-13. PubMed
- Sellmer L, Farschtschi S, Marangoni M, Heran MK, Birch P, Wenzel R, Friedman JM, Mautner VF. Non-optic glioma in adults and children with neurofibromatosis 1. Orphanet J Rare Dis. 2017; 12.; 34. PubMed
- Howell SJ, Hockenhull K, Salih Z, Evans DG. Increased risk of breast cancer in neurofibromatosis type 1: current insights. Breast Cancer (Dove Med Press). 2017; 9.; 531-536. PubMed
- Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, Blakeley J, Babovic-Vuksanovic D, Cunha KS, Ferner R, Fisher MJ, Friedman JM, Gutmann DH, Kehrer-Sawatzki H, Korf BR, Mautner VF, Peltonen S, Rauen KA, Riccardi V, Schorry E, Stemmer-Rachamimov A, Stevenson DA, Tadini G, Ullrich NJ, Viskochil D, Wimmer K, Yohay K, International Consensus Group on Neurofibromatosis Diagnostic Criteria (I-NF-DC)., Huson SM, Evans DG, Plotkin SR. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021; 23.; 1506-1513. PubMed
- Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR, COUNCIL ON GENETICS., AMERICAN COLLEGE OF MEDICAL GENETICS AND GENOMICS.. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics. 2019; 143.. PubMed
- Fisher MJ, Shih CS, Rhodes SD, Armstrong AE, Wolters PL, Dombi E, Zhang C, Angus SP, Johnson GL, Packer RJ, Allen JC, Ullrich NJ, Goldman S, Gutmann DH, Plotkin SR, Rosser T, Robertson KA, Widemann BC, Smith AE, Bessler WK, He Y, Park SJ, Mund JA, Jiang L, Bijangi-Vishehsaraei K, Robinson CT, Cutter GR, Korf BR, Neurofibromatosis Clinical Trials Consortium., Blakeley JO, Clapp DW. Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: a phase 2 trial. Nat Med. 2021; 27.; 165-173. PubMed
- Korfhage J, Lombard DB. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol Cancer Res. 2019; 17.; 1417-1428. PubMed