Epilepsi er en klinisk diagnose, som i høj grad er baseret på anamnesen
Videooptagelser på mobiltelefon er nyttige
EEG støtter diagnosen, men er ikke i sig selv diagnostisk
Behandling
Omfatter medicin, kirurgi, vagusstimulering og ketogen diæt
Henvisning
Ved mistanke om epilepsi, bør patienten henvises til pædiatrisk vurdering
Mistanke om West syndrom (infantile spasmer) bør føre til akut indlæggelse
Seneste væsentlige ændringer
Ingen
Diagnose
Baseres på sygehistorie og EEG-fund
Diagnosen kræver 2 sikre anfald adskilt af minimum 24 timer eller 1 anfald med høj sandsynlighed (mindst 60 %) for yderligere anfald eller diagnose af et epilepsisyndrom1
Der skelnes mellem fokale og generaliserede anfald. Ved fokale anfald starter den unormale elektriske aktivitet i et afgrænset område i hjernen, mens den ved generaliserede anfald starter i begge hjernehalvdele samtidig. Ved generaliserede anfald er der altid bevidsthedspåvirkning, mens fokale anfald kan kompliceres med bevidsthedspåvirkning
Barnet kan være rask mellem anfald. Anamnesen er meget vigtig, så forældreoplysninger er centrale
Videooptagelser af anfald med mobiltelefon er nyttige
Detaljeret anfaldsanamnese, udløsende faktorer, aura, barnets subjektive oplevelser under anfaldet, cyanose, toniske/kloniske kramper, afgang af urin eller afføring, slaphed, tidspunkt på døgnet
Graviditet, neonatalperiode, neurologisk udvikling og arvelige sygdomme
Epilepsityper
Godartede neonatale ikke familiære anfald
Selvbegrænsende kortvarige anfaldsfænomener, også kaldet femtedags-kramper
Omkring 7 % af neonatale krampeanfald er godartede, ikke-familiære neonatale anfald. Rammer lidt oftere drenge end piger
90 % af børnene har anfald mellem 4. og 6. levedøgn, resten mellem 1. og 7. levedøgn
Typisk ses serier af kloniske anfald ofte ensidige, men ikke sjældent med sideskift. Anfaldende ledsages i 1/3 af tilfældene af apnø. De
enkelte anfald i serien er kortvarige, og varer typisk 1-3 minutter
Forløb og prognose: Anfaldene ses hos i øvrigt raske spædbørn og varer typisk 20 timer, men kan vare op til 3 døgn. Herefter
forsvinder de af sig selv. Barnets udvikling vil i langt den overvejende del af tilfældene herefter være fuldstændig normal
Er en eksklusionsdiagnose, da de fleste neonatale anfald skyldes faktorer som asphyxi, blødning, infektion eller metaboliske forstyrrelser
West syndrom (tidligere infantile spasmer)
West syndrom er betegnelse for en samling af 3 symptomer: Epileptiske spasmer, karakteristiske forandringer på EEG (hypsarytmi) og negativ påvirkning af den kognitive udvikling
Debuterer typisk mellem 3 og 12 måneder, hyppigst omkring 5-månedersalderen. Anfald debuterer sjældent før 3-månedersalderen, og det er også sjældent, at de optræder første gang efter det første leveår
Epileptiske spasmer er pludselige, meget korte og ofte dobbeltsidige sammentrækninger i musklerne i arme, ben og den centrale del af kroppen. Oftest ses en sammenbukning af maven/hofteleddet med samtidig løft af skuldre og arme. Spasmerne kommer oftest i serier, og serierne kan forekomme mange gange i løbet af dagen
Årsager: Der er talrige årsaget til West syndrom af hvilke kan nævnes tuberøs sclerose, stofskiftelidelser og Down syndrom
Forløb og prognose: Afhængig af tilgrundliggende årsag. De fleste udvikler dog svær retardatio og eventuel autisme og ADHD lignende symptomer. Jo hurtigere der opnås anfaldsfrihed, jo bedre er prognosen. Der er en ikke helt ringe mortalitet (5 %) forbundet med West syndrom. Dette skyldes ikke anfaldene, men den tilgrundliggende lidelse
Dravet syndrom
Debuterer mellem 3 og 12 måneder, flest omkring 5-månedersalderen. Sjældent før 3-månedersalderen og efter første leveår. Udgør omkring 6 % af de epilepsier, der debuterer før 3-årsalderen
Årsag: Skyldes i langt de fleste tilfælde en nyopstået mutation i SCN1A-genet på kromosom 2. Kun hos 10 % af patienter med Dravet syndrom kan der ikke påvises en genetisk årsag
4 symptomer: Tidligt debuterende feberkramper (før 6 månedersalderen), myoklone ryk, atypiske absencer (f.eks. langvarige) og fokale anfald med påvirkning af bevidstheden
Forløb og prognose: Udviklingen sker i tre faser. Først er der en periode på 2-6 måneder med komplekse feberkramper og eventuel status epileptikus. Dernæst følger en periode med aggressive vanskeligt behandlelige anfald, som påvirker den kognitive udvikling i negativ retning. I sidste fase, der ofte begynder omkring 6-årsalderen, sker der en betydelig bedring i sværhedsgraden og hyppigheden af epileptiske anfald, men der vil fortsat være tale om betydelige kognitive, neurologiske og motoriske funktionsnedsættelser. Mortalitet er omkring 15 %
Rolandisk epilepsi
15-20 % af alle epilepsier hos børn
Debuterer i alderen 4-10 år. Anfald før 3 årsalderen og efter 14 årsalderen er ikke forenelige med diagnosen
Anfaldene kommer typisk under indsovning eller kort før opvågnen. Anfaldene er oftest enkeltstående anfald med ensidige sansemotoriske symptomer i ansigtet (hos 30 %), i mund/svælg (hos 50 %), manglende tale (hos 40 %) og betydelig savlen (hos 30 %). Det enkelte anfald varer 1-3 minutter. Hos omkring halvdelen af børnene kan man opleve, at et anfald udvikler sig til enten halvsidige krampeanfald eller generaliserede tonisk-kloniske anfald. Disse anfald kan følges af længerevarende (timer-dage) halvsidige lammelser, der forsvinder af sig selv
Under søvn har barnet epileptiform aktivitet, som kan ses på EEG
Forløb og prognose: Anfald ophører 3-4 år efter det første anfald, som regel før barnet fylder 16 år. De fleste har færre end 10 anfald og en mindre gruppe (omkring 10-20 %) har kun et enkelt anfald. En anden mindre gruppe (10-20 %) har mange anfald, men disse forsvinder også inden 16 årsalderen
Generaliserede tonisk/klononiske anfald
Akut bevidsthedstab med toniske efterfulgt af kloniske kramper
Eventuelt cyanose, tungebid og ekskretafgang
Anfald varer oftest mindre end 2 minutter
Efter anfald er patienterne postiktale, dvs. desorienterede og udmattede
Ofte muskeømhed efter anfald
Amnesi for anfaldet
God prognose. De fleste bliver anfaldsfrie efter nogle års behandling og kan trappes ud af behandling
Absenceepilepsi
Debut mellem 4. og 10. år, med hyppigste debuttidspunkt omkring 5-6-årsalderen. Det er usædvanligt, hvis absencer begynder før 4-årsalderen og efter 10-årsalderen
2 ud af 3 patienter er piger
Anfald varer typisk 10 - 40 sekunder. Barnet mister pludselig "tråden", hvilket eventuelt ledsages af øjenblink og øjendrejning. Herefter genoptager barnet sin aktivitet som om intet var hændt. Ofte mange anfald i døgnet
Anfaldene kommer oftest spontant, men kan fremprovokeres. En af de hyppigste provokerende faktorer er hyperventilation. Hvis ikke man kan fremprovokere et anfald hos et barn mistænkt for absenceepilepsi med hyperventilation, giver det grund til at revurdere diagnosen
Anfaldene ledsages af karakteristiske EEG* forandringer. Ved børneabsence-epilepsi er der desuden talrige generaliserede spike-waves (epileptiforme udladninger) i løbet af dagen – uden samtidige absencer
Forløb og prognose: mere end 60 % vokser fra epilepsien efter to år eller i løbet af teenageralderen. Mindre end 10% udvikler anden generaliseret epilepsi
Det er uhyre vigtig, at børneabsencer diagnosticeres og behandles, da uopdagede anfald kan have betydelige konsekvenser for barnets indlæring og lede til fejldiagnostik som f.eks. koncentrationsvanskeligheder
Juvenil absenceepilepsi
Debuterer i 8-20-årsalderen typisk i 9-13-årsalderen
Typiske absencer, som er længere og ikke forekommer så hyppigt (ikke dagligt) som ved børneabsence epilepsi. Bevidsthedstabet kan være totalt eller delvis og er ikke så markant som ved børneabsenceepilepsi. Patienten kan i nogle tilfælde svare på spørgsmål, som bliver stillet under et anfald
Over 90 % har også generaliserede tonisk-kloniske anfald, som dog oftest ses, hvis patienten ikke er velbehandlet. Hos 14-27 % opleves der generaliserede tonisk-kloniske anfald, før absencerne debuterer. Nogle får myoklonier under anfald
Forløb og prognose: 70-80 % bliver anfaldsfrie på behandlingen, men recidiverer ofte ved udtrapning af medicinen, hvorfor behandlingen er livslang
Nogle har indlæringsvansleligheder og/eller ADHD
Juvenil myoklon epilepsi
3 symptomer: absencer, myoklone ryk ved opvågning og generaliserede tonisk/kloniske anfald. Udgør omkring 8% af børneepilepsier
Debuterer typisk mellem 14 og 25 år
Den dominerende anfaldstype er myoklone ryk, som ses hos alle patienter med JME. De myoklone ryk ses typisk ½-1 time efter opvågning. De kan være ganske beskedne, men kan medføre ufrivillige bevægelser som gør, at patienten vælter ting, eller de kan være så voldsomme, at patienten falder. De kan være symmetriske eller asymmetriske. Et fåtal (<10 %) har myoklone ryk som eneste anfaldstype. Mere end 90 % har generaliserede tonisk-kloniske anfald, typisk i forbindelse med opvågning. Absencer, der ses hos omkring 1/3. De er ofte kortere og mindre hyppige, end det ses ved børneabsence epilepsi og juvenil absence epilepsi
Forløb og prognose: De fleste responderer godt på behandling og bliver anfaldsfrie. Dog oftest recidiv ved forsøg på behandlingsophør, så behandlingen vil oftest være livsvarig
Starter med myoklone ryk og derefter atonisk anfald, hvor alle muskler bliver slappe, og patienten falder. Eventuelt tonisk/klonisk krampeanfald efterfølgende
Forløb og prognose: Op til 80 % opnår fuld anfaldskontrol indenfor 3 år. Nogen udvikler sig efterfølgende normalt men ikke sjældent ses mental retardering
Andre sjældnere epilepsiformer i barnealderen
Omfatter blandt andre Lennox-Gasteau syndrom, Landau kleffner og Oktahara Syndrom
Supplerende undersøgelser i almen praksis
Patienter med anfald som er overstået, henvises elektivt eventuelt med samtidig bestilling af EEG
Ved mistanke om West syndrom (infantile spasmer) dog akut henvisning
Specialistudredning
EEG
Evt. med provokationer som flimmerlys, søvn og hyperventilering
Foretages efter andet afebrile krampeanfald eller ved mistanke om andre anfaldsfænomener end grand mal
Normalt EEG udelukker ikke epilepsi, og 3 - 5 % af raske børn har klassisk epileptiform aktivitet på EEG2
Langtids video-EEG bør foretages ved uafklarede anfald, både når der er tvivl om klassifikation af epileptiske anfald og syndromer, og når der er mistanke om non-epileptiske anfald3
Rolandisk epilepsi
Har karakteristiske centrotemporale spike-fokus
Absenceepilepsi
Har 3/sek spike-wave komplekser
West syndrom (infantile spasmer)
Har kontinuerlige uorganiserede multifokale brede høje bølger, kaldt hypsarytmi
MR/CT
Bør foretages ved epilepsidebut, med mindre klinik og EEG tillader en af diagnoserne rolandisk epilepsi, børneabsenceepilepsi, juvenil absenceepilepsi eller juvenil myoklon epilepsi3
EKG (eventuelt)
For at udelukke rytmeforstyrrelser. Ved bevidsthedstab under fysisk aktivitet bør patienten vurderes af børnekardiolog
Elektrolytforstyrrelser som hypokalcæmi, hypo/-hypernatriæmi eller hypo-/hyperglykæmi
Neonatale kramper er oftest forårsaget af organiske skader eller metaboliske sygdomme, hvilket vil sige at godartede ikke familiære neonatale anfald er en eksklusionsdiagnose
Hjerterytmeforstyrrelser som f.eks. forlænget QT-syndrom. Optræder bevidsthedstab under fysisk aktivitet, bør barnet vurderes af børnekardiolog2
Behandling
Behandlingsmål
At skabe forudsætninger for at barnet får et godt liv ved at reducere anfaldsfrekvens og komplikationer
Generelt om behandlingen
Korrekt klassificering er vigtig af hensyn til valg af behandlingsstrategi
Medikamentel terapi, men vurder også ikke-medikamentelle faktorer som
Psykiske
Pædagogiske
Sociale forhold
Kirurgi
Hvis der ikke opnås anfaldskontrol med medikamenter, bør man efter 1-2 år overveje udredning med henblik på epilepsikirurgi3
Ketogen diæt
Psykiske problemer
Epilepsi er en uforudsigelig sygdom, som pludseligt kan give anfald. Dette disponerer til psykiske problemer, som kan kræve behandling
Multidisciplinær håndtering
Alle børn med farmakoresistente anfald bør gennemgå en tværfaglig multidisciplinær diagnostisk evaluering
Håndtering i almen praksis
Børn mistænkt for epilepsi bør henvises til pædiatrisk vurdering
Råd til patienten
Efterleve den medikamentelle behandling
Aldrig svømme alene og ikke kravle i træer
Medicinsk behandling
Der er ikke holdepunkt for opstart af medikamentel behandling efter kun ét uprovokeret krampeanfald4
Tidlig antiepileptisk behandling påvirker ikke langtidsprognosen (undtagelse West syndrom)4
Overvej seponering af antiepileptisk behandling efter 2 års anfaldsfrihed hos børn og unge under 18 år med epilepsi af både strukturel og ikke strukturel årsag3
Omkring 50 % bliver anfaldsfri ved hjælp af ét medikament
Man skal altid afprøve medikamentet til adækvat serumniveau eller bivirkninger, før man afskriver præparatet
Det er også vigtigt at være opmærksom på, at medikamenter kan give paradoksale effekter og forværre anfald
Dette gælder specielt hvis generaliserede anfald, som absencer, bliver mistolket som fokale anfald, og derved fører til forkert medikamentvalg
Mest aktuelt hos børn med psykisk udviklingshæmning og epileptiske encefalopatier
Til generaliserede epilepsier er valproat førstevalg. Imidlertid frarådes valproat til piger/kvinder i den fødedygtige alder pga. risiko for teratogen effekt og for udvikling af polycystisk ovariesyndrom. Lamotrigin synes at være noget mindre effektivt end valproat mod absencer, men præparatets bivirkningsprofil er til gengæld mere attraktiv. Lamotrigin kan forværre myoklonier. Af hensyn til bivirkningsprofilen hos piger kan bl.a. også levetiracetam afprøves før valproat. Ethosuximid virker ikke mod generaliserede tonisk kloniske anfald, men er velegnet til absencer.
Ved fokal epilepsi bør lamotrigin vælges. Hos børn < 2 år kan det være vanskeligt at opnå tilstrækkeligt højt plasmaniveau af det ellers velegnede oxcarbazepin pga. autoinduktion. Ved børneepilepsi med centrotemporale spikes (Rolandisk epilepsi) kan man, hvis der kun har været 1-2 anfald og i samarbejde med barn og familie, vælge at afvente evt. medicinsk behandling. Ved hyppige anfald vælges oftest valproat, levetiracetam kan også anvendes. Oxcarbazepin og lamotrigin kan forværre denne epilepsiform. Sultiam kan være velegnet, men kræver udleveringstilladelse
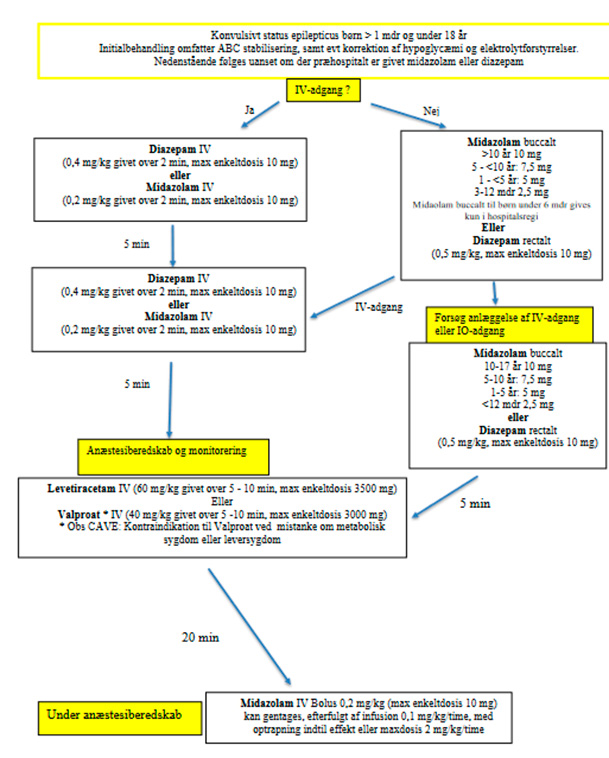
Overvej at behandle epileptiske anfald, der varer længere end 3-5 minutter, med buccal midazolam frem for rektal diazepam, idet der er lavere risiko for recidiv ved brug af buccal midazolam3
Status epilepticus
Definitioner
Status epilepticus: En tilstand med epileptiske anfald af abnorm lang varighed, som efter et tidsrum medfører risiko for permanent cerebral skade
Konvulsivt status epileptikus: Langvarigt bilateralt (generaliseret) tonisk-klonisk anfald > 5 min. eller gentagne bilaterale (generaliserede) tonisk-kloniske anfald, hvor patienten ikke genvinder fuld bevidsthed imellem anfaldene
Non-konvulsivt status epileptikus: En heterogen gruppe af tilstande med længerevarende epileptiske anfald (>10 min) med bevidsthedspåvirkning uden prominente motoriske symptomer. Diagnosen dokumenteres med EEG
Andre former for status epileptikus: Andre former for SE omfatter f.eks. myoklon SE eller fokal motorisk SE uden bevidsthedspåvirkning (epilepsia partialis continua)
Konvulsivt status er en livstruende tilstand. Der er risiko for senfølger og tilstanden skal behandles aggresivt efter nedenstående retningslinje:
Udredning og behandling af medicinsk intraktabel epilepsi hos børn og unge3
Ved medicinsk intraktabel epilepsi kan overvejes udredning med henblik på epilepsikirurgi, ketogen diæt eller nervus vagus stimulator
Det er i dag muligt i udvalgte tilfælde at operere børn med medicinsk intraktabel epilepsi, hvilket har forbedret prognosen både hvad angår anfald og hvad angår kognitiv udvikling
Hvis kirurgi ikke er egnet, kan ketogen diæt overvejes
Er ketogen diæt uden effekt eller ikke gennemførligt kan vagus-stimulation overvejes
Epilepsikirurgi
Læsionsektomi
Kallosotomi
Ved myoklon-astatisk epilepsi, hvor man fjerner de forreste 2/3 af corpus callosum
Resektionskirurgi ved fokale epilepsier
Excision af det anfaldsgivende område efter lokalisering med MR og EEG
Kirurgen kan fra at fjerne et mindre anfaldsgivende hjerneområde foretage en funktionel hemisfærektomi
Vagusnervestimulering
Kan være et godt behandlingsalternativ hos børn med farmakoresistent epilepsi
Det er få bivirkninger, og disse bliver med tiden mindre generende
Vagusnervestimulering kan have effekt ved mange forskellige anfaldsformer, og særligt ved atoniske anfald
Forebyggende behandling
Undgå risikofaktorer
Søvndeprivation
Flimmerlys (kun relevant hvis EEG viser fotosensibilitet)
Alkohol
Henvisning
Ved mistanke om epilepsi bør patienten henvises til pædiatrisk vurdering
Langvarige anfald (>10 min) eller mistanke om West syndrom (infantile spasmer) bør føre til akut indlæggelse
Opfølgning
Plan
Individuel
Hvad bør man kontrollere
Effekt af behandling
Vækst og udvikling
Sociale, psykologiske, neurologiske og somatiske færdigheder
Bivirkninger af medikamenter, specielt kognitiv påvirkning
Sygdomsforløb
Varierer afhængig af epilepsitype og eventuel underliggende lidelse
Komplikationer
Symptomer som koncentrationsbesvær, perceptionsforstyrrelser, emotionelle forstyrrelser og mental retardation vil accentueres hos et barn med epilepsi
Status epilepticus, definitionsmæssigt langvarigt epileptisk anfald eller multiple anfald uden at genvinde bevidstheden
Cerebrale bivirkninger af medicin
Hjerneskade på grund af langvarige anfald >10-15 min
Psykisk sygdom som følge af overbeskyttelse og følelse af manglende selvkontrol, som de epileptiske anfald ofte fremkalder
Patienter med epilepsi har sammenlignet med baggrundspopulationen en vis overdødelighed. Risikofaktorer omfatter strukturel eller metabolisk ætiologi, hyppighed af kramper samt natlige kramper. Børn med ukompliceret epilepsi er ikke i risiko for epilepsirelateret død4
Prognose
Prognosen er helt afhængig af epilepsi-type samt af underliggende årsager
Generelt
Mange epilepsier som debuterer i barneårene er godartede, og 50 % af børnene har ikke epilepsi i voksenalder
Nogle børneepilepsier er imidlertid meget alvorlige. Næsten alle alvorlige, generaliserede epilepsier debuterer i tidlig barnealder
Rolandisk epilepsi
Prognosen er god ved Rolandisk epilepsi, hvor de fleste anfald ophører før 20-årsalderen
Absenceepilepsi
80 % bliver anfaldsfrie før 20-årsalderen
West syndrom (Infantile spasmer)
50 % fortsætter med at have anfald
30 % udvikler Lennox-Gastauts syndrom
85-90 % udvikler psykomotorisk retardation
Baggrundsoplysninger
Definition
Epilepsi er en tilstand med gentagne spontane anfald forårsaget af elektriske forstyrrelser i hjernen
Epileptisk anfald
Uprovokerede, pludselige motoriske, sensoriske eller psykiske fænomener forårsaget af en forbigående forstyrrelse af dele eller hele hjernen som følge af elektrisk udladning af hyperexcitable nerveceller
Anfaldene er forskellige fra person til person, men er som oftest stereotype for en given patient
Epileptisk syndrom
Er en tilstand, hvor en række symptomer og tegn forekommer samtidigt
Det inkluderer anfaldstype(r), EEG-fund, anatomisk fokus, anfaldsprovokerende faktorer, alder ved debut, sværhedsgrad og kronicitet
Er højest i de to første leveår og falder siden gennem opvæksten
Den kumulative risiko for at få epilepsi som barn (0-15 år) er ca. 1 %
40 % af al epilepsi debuterer i barndommen
Prævalensen
Stiger med stigende alder
Prævalensen i barnealderen er 4-5 pr. 1000
Anfaldstype
0-9 år
Incidensen af generaliserede anfald er højere end af fokale anfald
10-19 år
Fokale og generaliserede anfald er lige hyppige
Ætiologi og patogenese
Meget ofte finder man ingen årsag
Ætiologien bag et epileptisk syndrom kan variere
Ikke alle former for epilepsi hos børn kan klassificeres inden for de epileptiske syndromer, men når dette er muligt, kan man bedre vælge adækvate undersøgelser, korrekt behandling og forudsige prognosen
Fysiske årsager
Traume
Tumor cerebri
Tuberøs sklerose
Hypertension og vaskulære katastrofer
Malformationer i CNS
Cerebral parese
Genetiske defekter
Andre årsager til kramper
Hypo- og hyperglykæmi
Hypoksi og asfyksi
Meningitis og meningoencephalitis
Uræmi
Hypo- og hypernatriæmi
Hypokalcæmi
Fenylketonuri
ICPC-2
ICD-10/SKS-koder
Patientinformation
Hvad du bør informere patienten om
Information om sygdommens art og vejledning i erhvervsvalg vil være relevant i ungdomsårene
Professor, ph.d., praktiserende læge, Institut for Folkesundhed - Almen Medicin, Aarhus Universitet
Har du en kommentar til artiklen?
Bemærk venligst, at du IKKE vil modtage svar på henvendelser, der omhandler din egen sygdom, pårørendes sygdom, blodprøvesvar, hjælp til at udarbejde skoleopgaver og litteratursøgning.